
A two-way ANOVA test is a statistical test used to determine the effect of two nominal predictor variables on a continuous outcome variable. A two-way ANOVA tests the effect of two independent variables on a dependent variable. This blog post contains the following steps of two-way ANOVA in r:
- Introduction
- Repeated measures
- Randomized blocks
- Import data
- Format variables
- Fit two way analysis of variance model
- Compute Tukey Honest Significant differences
- Compute mean and standard error
- Visualize effects
Introduction to two-way ANOVA
We can use a two-way analysis of variance when we have one measurement variable and two nominal variables. This method analyzes data in which every value of one nominal variable appears in combination with each value of the other nominal variable. It tests three null hypotheses.
H0: All means of genotypes are equal
H1: At least two means of genotypes are unequal
The means of observations grouped by the other factor are the same.
H0: All means of gender are equal
H1: At least two means of gender are unequal
There is no interaction between the two factors. The interaction test tells you whether the effects of one factor depend on the other factor.
H0: There is no interaction between genotype and gender
H1: There is significant interaction
When the interaction term is significant, the usual advice is that you should not test the effects of the individual factors.Repeated measures
One experimental design that people analyze with a two-way ANOVA is repeated measures. In this design, the observation has been made on the same individual more than once. This usually involves measurements taken at different time points or at different places. Repeated measures experiments are often done without replication, although they could be done with replication.
Randomized blocks
Another experimental design that is analyzed by a two-way ANOVA is randomized blocks. This often occurs in agriculture, where you may want to test different treatments on small plots within larger blocks of land. Because the larger blocks may differ in some way that may affect the measurement variable, the data are analyzed with a two-way ANOVA, with the block as one of the nominal variables. Each treatment is applied to one or more plots within the larger block, and the positions of the treatments are assigned at random. Here, the example used shows the enzyme activity of mannose-6-phosphate isomerase and MPI genotypes in the amphipod crustacean. Amphipods were separated by gender to know whether the gender also affected enzyme activity.
Import data
I often recommend to first clear all the objects or values in the global environment using rm(list = ls(all = TRUE)) before importing the data set. You can also clear the plots using graphics.off() and clear everything in console using shell() function.
rm(list = ls(all = TRUE))
graphics.off()
shell("cls")Now let’s import the data set using read.csv() function. I have already saved the data file as CSV (comma delimited file) in the working directory. The file argument specify the file name with extension CSV. In header argument you can set a logical value that will indicate whether the data file contains first variable names as first row. In my data set the file contains the variable names in the first row, so I shall use TRUE for this argument. The head() function will print the first six rows of the data set.
data <- read.csv(file = "data_2way.csv",
header = TRUE)
head(data)# genotype gender activity
# 1 FF Female 3.39
# 2 FF Female 3.34
# 3 FF Female 3.59
# 4 FO Female 3.58
# 5 FO Female 4.12
# 6 FO Female 4.72Format variables
The str() or structure-function will tell us the format for each column in the data frame. It gives information about rows and columns. It also gives information on whether the variables are being read as integer, factor or number.
In some cases, we may have a variable coded as a character or integer that we would like R to recognize as a factor variable. For this purpose as.factor() function is used. In our data file, the genotype and gender are being read as character in R instead of being recognized as factor variables. To change it to factor assign the function of as.factor() where x argument represents the factor component of the data set (data$genotype).
This will change the structure of the variable genotype and R will recognize it as a factor. Similarly, assign the same function for the second variable by replacing genotype with gender in the previous command. Now using again the str() function will show the second variable is being read as a factor.
str(data)# 'data.frame': 18 obs. of 3 variables:
# $ genotype: Factor w/ 3 levels "FF","FO","OO": 1 1 1 2 2 2 3 3 3 1 ...
# $ gender : Factor w/ 2 levels "Female","Male": 1 1 1 1 1 1 1 1 1 2 ...
# $ activity: num 3.39 3.34 3.59 3.58 4.12 4.72 5.06 4.05 4.09 2.2 ...data$genotype <- as.factor(x = data$genotype)
data$gender <- as.factor(x = data$gender)
str(data)# 'data.frame': 18 obs. of 3 variables:
# $ genotype: Factor w/ 3 levels "FF","FO","OO": 1 1 1 2 2 2 3 3 3 1 ...
# $ gender : Factor w/ 2 levels "Female","Male": 1 1 1 1 1 1 1 1 1 2 ...
# $ activity: num 3.39 3.34 3.59 3.58 4.12 4.72 5.06 4.05 4.09 2.2 ...Fit two-way ANOVA model
First attach the data set to the R search path using attach() function. This means that the database is searched by R when evaluating a variable, so objects in the database can be accessed by simply giving their names. This will help us to reduce the argument data$genotypes to genotypes only.
We can access the functions of two way analysis of variance using R stats package. Load this package using the library() function before fitting the two way analysis of variance model. The two way analysis of variance model can be fitted in two ways as per the objectives or to get the information a researcher is most interested in. If you want to see the main effects without interaction then fit analysis of variance model using aov() function.
This function requires formula as its argument to represent what output you wants to get from it. For main effects model use the response variable (activity in this case) separated by genotype and gender. In this model formula the response variable is separated from factor variables using tilde operator ~. The factor variables are combined using the plus sign. To print this model use anova() function for this object.
Now let’s fit the same model including the interaction effect. Use the same command as previously used to fit main effects by combining the variable factors with an additional argument genotype:gender representing the interaction term.
attach(data)
library(stats)
# Main effect
aov.res1 <- aov(activity ~
genotype +
gender)
# Interaction effect
aov.res2 <- aov(activity ~
genotype +
gender +
genotype:gender)
# OR
aov.res2 <- aov(activity ~
genotype*gender)
anova(aov.res1)# Analysis of Variance Table
#
# Response: activity
# Df Sum Sq Mean Sq F value Pr(>F)
# genotype 2 6.2525 3.12627 10.4111 0.001698 **
# gender 1 1.3833 1.38334 4.6068 0.049855 *
# Residuals 14 4.2039 0.30028
# ---
# Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1anova(aov.res2)# Analysis of Variance Table
#
# Response: activity
# Df Sum Sq Mean Sq F value Pr(>F)
# genotype 2 6.2525 3.12627 10.9923 0.001938 **
# gender 1 1.3833 1.38334 4.8640 0.047669 *
# genotype:gender 2 0.7911 0.39554 1.3908 0.286269
# Residuals 12 3.4129 0.28441
# ---
# Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Enzyme activity is significantly affected by the both factor variables individually. However, this effect was non-significant for interaction term.Compute Tukey Honest Significant differences
I shall use TukeyHSD() to compute the significant differences among the mean values. This test creates a set of confidence intervals on the differences between the means of the levels of a factor with the specified family-wise probability of coverage. The intervals are based on the Studentized range statistic, Tukey’s ‘Honest Significant Difference’ method.
This test requires certain arguments. The x argument specifies the fitted model object (aov.res1 in this case), usually an aov() fit. The argument ordered specify a logical value indicating if the levels of the factor should be ordered according to increasing average in the sample before taking differences. If ordered is set to TRUE then the calculated differences in the means will all be positive. The significant differences will be those for which the lower endpoint is positive.
The argument which specify a character vector listing terms in the fitted model for which the intervals should be calculated. Default value specifies all the terms. You can also specify the variable factor name for this argument. For interaction use ”genotype:gender” as the value for which argument. For confidence level use conf.level argument to set its value. The default value for this argument is 0.95 representing the 5% level of significance.
# Mean comparison test for genotypes
TukeyHSD(x = aov.res1,
ordered = FALSE,
which = "genotype",
conf.level = 0.95)# Tukey multiple comparisons of means
# 95% family-wise confidence level
#
# Fit: aov(formula = activity ~ genotype + gender)
#
# $genotype
# diff lwr upr p adj
# FO-FF 0.7866667 -0.04137865 1.614712 0.0635754
# OO-FF 1.4416667 0.61362135 2.269712 0.0012185
# OO-FO 0.6550000 -0.17304532 1.483045 0.1322481# Mean comparison test for gender
TukeyHSD(x = aov.res1,
ordered = FALSE,
which = "gender",
conf.level = 0.95)# Tukey multiple comparisons of means
# 95% family-wise confidence level
#
# Fit: aov(formula = activity ~ genotype + gender)
#
# $gender
# diff lwr upr p adj
# Male-Female -0.5544444 -1.108486 -0.0004029292 0.049855# Mean comparison test for interaction (genotype:gender)
TukeyHSD(x = aov.res2,
ordered = FALSE,
which = "genotype:gender",
conf.level = 0.95)# Tukey multiple comparisons of means
# 95% family-wise confidence level
#
# Fit: aov(formula = activity ~ genotype * gender)
#
# $`genotype:gender`
# diff lwr upr p adj
# FO:Female-FF:Female 0.7000000 -0.7625920 2.1625920 0.6090747
# OO:Female-FF:Female 0.9600000 -0.5025920 2.4225920 0.3028696
# FF:Male-FF:Female -0.9333333 -2.3959253 0.5292586 0.3288700
# FO:Male-FF:Female -0.0600000 -1.5225920 1.4025920 0.9999909
# OO:Male-FF:Female 0.9900000 -0.4725920 2.4525920 0.2754196
# OO:Female-FO:Female 0.2600000 -1.2025920 1.7225920 0.9892495
# FF:Male-FO:Female -1.6333333 -3.0959253 -0.1707414 0.0258232
# FO:Male-FO:Female -0.7600000 -2.2225920 0.7025920 0.5305000
# OO:Male-FO:Female 0.2900000 -1.1725920 1.7525920 0.9825802
# FF:Male-OO:Female -1.8933333 -3.3559253 -0.4307414 0.0094477
# FO:Male-OO:Female -1.0200000 -2.4825920 0.4425920 0.2498774
# OO:Male-OO:Female 0.0300000 -1.4325920 1.4925920 0.9999997
# FO:Male-FF:Male 0.8733333 -0.5892586 2.3359253 0.3927173
# OO:Male-FF:Male 1.9233333 0.4607414 3.3859253 0.0084220
# OO:Male-FO:Male 1.0500000 -0.4125920 2.5125920 0.2262177Compute mean and standard error
Before visualizing main and interaction effects first compute mean values and standard error. Load the library dplyr that states the grammar of data manipulation. Connect data set to group it by first genotypes and then summarise it to get mean values and standard error using group_by() and summarise(), respectively. For connection, we shall use %>% pipe operator. In function summarise() set functions for the objects avg_A and se to compute mean and standard error values. Print function will display the results for this object.
Similarly, get mean values and standard error for second variable factor using the same command except connecting the data set to group_by() gender using %>% pipe operator. Print function for this object will display the results. Again use the same command for the interaction term. Here you will connect the data set to group_by() genotype and gender using the %>% pipe operator. Print function for this object will display the results. Attach these three objects to access its components by simply giving their names while visualizing plots.
library(dplyr)
# For main effects
MeanSE_A = data %>%
group_by(genotype) %>%
summarise(avg_A = mean(activity),
se = sd(activity)/sqrt(length(activity)))
MeanSE_B = data %>%
group_by(gender) %>%
summarise(avg_B = mean(activity),
se = sd(activity)/sqrt(length(activity)))
print(MeanSE_A)# # A tibble: 3 x 3
# genotype avg_A se
# <fct> <dbl> <dbl>
# 1 FF 2.97 0.223
# 2 FO 3.76 0.240
# 3 OO 4.42 0.281print(MeanSE_B)# # A tibble: 2 x 3
# gender avg_B se
# <fct> <dbl> <dbl>
# 1 Female 3.99 0.198
# 2 Male 3.44 0.326# For interaction term
MeanSE_AB = data %>%
group_by(genotype, gender) %>%
summarise(avg_AB = mean(activity),
se = sd(activity)/sqrt(length(activity)))
print(MeanSE_AB)# # A tibble: 6 x 4
# # Groups: genotype [3]
# genotype gender avg_AB se
# <fct> <fct> <dbl> <dbl>
# 1 FF Female 3.44 0.0764
# 2 FF Male 2.51 0.157
# 3 FO Female 4.14 0.329
# 4 FO Male 3.38 0.186
# 5 OO Female 4.40 0.330
# 6 OO Male 4.43 0.535attach(MeanSE_A)
attach(MeanSE_B)
attach(MeanSE_AB)Visualize effects in two-way ANOVA
Plot genotype
Let’s start to display the main and interaction effects using grammar of graphics function. This will require to load the package ggplot2. Create an object p1 using the grammar of graphics function. This function requires data set (MeanSE_A) and some aesthetic mappings to use for the plot. In aesthetic mappings function the argument x specify the variable genotype and argument y specify mean values (avg_A). Print function will print a blank plot showing x-axis and y-axis labels.
Now let’s add layers to this plot object. For plotting bars use geom_bar() function. In stat argument set the value as identity to leave the data as is. In color argument specify the color for borders of the bars. The position argument specify the Position adjustment, either as a string, or the result of a call to a position adjustment function. The argument width specify the width of the bars. Printing this layer with plot object will display bars on the plot.
Let’s add the second layer to show error bars on the bars of the plot. For this use geom_errorbar() function. In aesthetic mappings you can specify the maximum ymax and minimum ymin values to show the heights of the error bars. Use the same position argument as used in previous command so that the error bars could place in the center of the bars. You can adjust width of the error bars by setting the value for width argument.
Let’s change the main title, X and Y axis labels of the plot. For this use labs() function. In title you can specify the main title of the plot. For X and Y argument specify the labels to be used for X and Y axis.
library(ggplot2)
# Create plot object
p1 = ggplot(MeanSE_A, aes(x = genotype,
y = avg_A))
# Adding layers to plot object
# Plotting bars
plotA = p1 +
geom_bar(stat = "identity",
color = "black",
position = position_dodge(width=0.9),
width = 0.8)
# Adding error bars
plotB = plotA +
geom_errorbar(aes(ymax = avg_A + se,
ymin = avg_A - se),
position = position_dodge(width=0.9),
width = 0.25)
# Changing main title, X & Y labels
plotC = plotB +
labs(title = "",
x = "genotype",
y = "MPI Activity")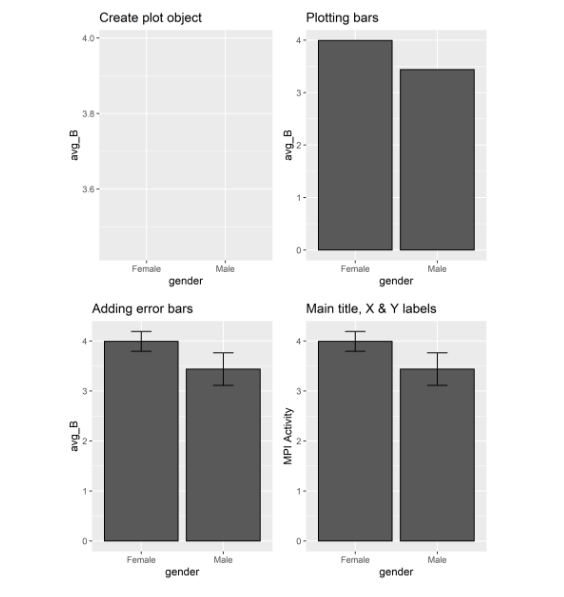
Plot gender
The same functions will be used to plot bar graph for second factor. For plotting gender first create an object p2 using grammar of graphics function. Use same arguments in this object changing the values to represent the second factor data set (MeanSE_B). Similarly, use gender for x argument and avg_B for y argument Print function will display the empty plot for this object just showing the X and Y axis labels.
For adding layers to this plot object use the same functions as used in adding layers for first variable factor. Just change the values of the arguments representing factor B instead of factor A. This will result in printing all the layers to the object plot.
# Create plot object
p2 = ggplot(MeanSE_B, aes(x = gender,
y = avg_B))
# Adding layers to p2 object
# Adding bars
plotA = p2 +
geom_bar(stat = "identity",
color = "black",
position = position_dodge(width=0.9))
# Adding error bars
plotB = plotA +
geom_errorbar(aes(ymax = avg_B + se,
ymin = avg_B - se),
position = position_dodge(width=0.9),
width = 0.25)
# Changing main title, X and Y labels
plotC = plotB +
labs(title = "",
x = "gender",
y = "MPI Activity")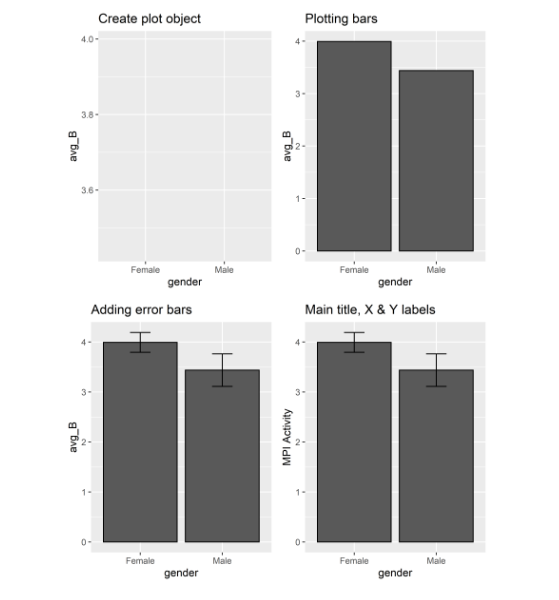
Plot interaction
First create the plot object similar to the one created for main effects. However, you will change the arguments to represent the interaction data set (MeanSE_AB). In aesthetic mappings define X, Y and fill arguments. The argument X specify gender or the variable name that you want to place on X axis. In Y argument set the mean values for interaction term. The argument fill specify the second factor used to set the color and fill of geom elements on a plot. I shall set the genotype for this argument. Print function will display the empty plot for this object just showing the X and Y axis labels.
Now add the layers to this plot object. Use same arguments for geom_bar() as used in previous command to plot bars except replacing the values of arguments to represent the interaction term instead of main effects. Now I shall type a different layer for adding color for fill and changing legend text. For this use scale_fill_manual() function. In values argument set the fill color for the bars. In labels you can specify labels for the legend text. Print function for this layer will add fill color and labels for legend text.
To add error bars on top of the plot bars use geom_errorbar() function. You can specify settings for aesthetic mappings (aes()), position and width of the error bars. In aesthetic mapping set the maximum ymax and minimum ymin values for error bars on Y axis. I shall set these values by adding avg_AB + se and subtracting avg_AB - se standard error from height of the bars. Specify position argument same as used in previous command (command for plotting bars geom_bar()). This will result placing the error bars exactly on the same position as the bars were plotted. You can change the width of the error bars by setting the value for width argument. Printing this layer will add error bars.
library(ggplot2)
# Create plot object
p3 = ggplot(MeanSE_AB, aes(x = gender,
y = avg_AB,
fill = factor(genotype)))
# Adding layers to plot object
# Plotting bars
plotA = p3 +
geom_bar(stat = "identity",
color = "black",
position = position_dodge(width=0.9))
# Adding color for fill and changing legend text
plotB = plotA +
scale_fill_manual(values = gray(1:3/3),
labels = c("FF", "FO", "OO"))
# Adding error bars
plotC = plotB +
geom_errorbar(aes(ymax = avg_AB + se,
ymin = avg_AB - se),
position = position_dodge(width=0.9),
width = 0.25)
# Changing main title, X and Y labels and legend title
plotD = plotC +
labs(title = "",
x = "gender",
y = "MPI Activity",
fill = "genotype")
print(p3 + labs(title = "Create plot object"))Conclusion
In summary, the Two-Way ANOVA is a powerful statistical technique used to examine the influence of two independent categorical variables on a single continuous dependent variable. It not only assesses the main effects of each factor individually but also determines whether there is a significant interaction effect between them showing whether the impact of one factor depends on the level of the other. By partitioning variance and comparing group means, Two-Way ANOVA provides deeper insight into complex datasets where multiple variables may jointly influence outcomes. Overall, it is an essential tool for researchers seeking to understand patterns, relationships, and combined effects within experimental and observational studies. Data Science Blog